Medication reference
Sorafenib
Kinase Inhibitor [EPC] — ORAL
Sorafenib — Kinase Inhibitor [EPC]. INDICATIONS AND USAGE Sorafenib is a kinase inhibitor indicated for the treatment of • Unresectable hepatocellular carcinoma ( 1.1 ) • Advanced renal
Brand names
sorafenib
Active ingredients
SORAFENIBSORAFENIB TOSYLATE
Indications
INDICATIONS AND USAGE Sorafenib is a kinase inhibitor indicated for the treatment of • Unresectable hepatocellular carcinoma ( 1.1 ) • Advanced renal cell carcinoma ( 1.2 ) • Locally recurrent or metastatic, progressive, differentiated thyroid carcinoma (DTC) refractory to radioactive iodine treatment ( 1.3 ) 1.1 Hepatocellular Carcinoma Sorafenib tablets are indicated for the treatment of patients with unresectable hepatocellular carcinoma (HCC). 1.2 Renal Cell Carcinoma Sorafenib tablets are indicated for the treatment of patients with advanced renal cell carcinoma (RCC). 1.3 Differentiated Thyroid Carcinoma Sorafenib tablets are indicated for the treatment of patients with locally recurrent or metastatic, progressive, differentiated thyroid carcinoma (DTC) that is refractory to radioactive iodine treatment.
Dosage
DOSAGE AND ADMINISTRATION The recommended dosage is 400 mg orally twice daily without food. ( 2.1 ) 2.1 Recommended Dosage The recommended dosage of sorafenib tablets is 400 mg orally twice daily without food (at least 1 hour before or 2 hours after a meal) until the patient is no longer clinically benefiting from therapy or until unacceptable toxicity. 2.2 Dosage Modifications for Adverse Reactions Recommended Dosage Modifications The recommended dosage modifications for adverse reactions are provided in Tables 1, 2, and 3. Table 1: Recommended Dose Reductions for Adverse Reactions Dose Reduction Hepatocellular Carcinoma and Renal Cell Carcinoma Differentiated Thyroid Carcinoma First Dose Reduction 400 mg orally once daily 400 mg orally in the morning and 200 mg orally in the evening about 12 hours apart OR 200 mg orally in the morning and 400 mg orally in the evening about 12 hours apart Second Dose Reduction 200 mg orally once daily OR 400 every other day 200 mg orally twice daily Third Dose Reduction None 200 mg orally once daily Table 2: Recommended Dosage Modifications of Sorafenib Tablets for Adverse Reactions Adverse Reaction Severity 1 Sorafenib Tablets Dosage Modification Cardiovascular Events [see Warnings and Precautions ( 5.1 )] Cardiac Ischemia and/or Infarction Grade 2 and above Permanently discontinue. Congestive Heart Failure Grade 3 Interrupt 2 until Grade 1 or less, resume at reduced dose by 1 dose level. 3 Grade 4 Permanently discontinue. Hemorrhage [see Warnings and Precautions ( 5.2 ) ] Grade 2 and above requiring medical intervention Permanently discontinue. Hypertension [see Warnings and Precautions ( 5.3 )] Grade 2 (symptomatic/persistent) OR Grade 2 symptomatic increase by greater than 20 mm Hg (diastolic) or greater than 140/90 mm Hg if previously within normal limits OR Grade 3 Interrupt until symptoms resolve and diastolic blood pressure less than 90 mm Hg, then resume at reduced dose by 1 dose level. 3 If needed, reduce another dose level. 3 Grade 4 Permanently discontinue. Gastrointestinal Perforation [see Warnings and Precautions ( 5.5 )] Any grade Permanently discontinue. QT Interval Prolongation [see Warnings and Precautions ( 5.9 )] Greater than 500 milliseconds OR Increase from baseline of 60 milliseconds or greater Interrupt and correct electrolyte abnormalities (magnesium, potassium, calcium). Use medical judgement before restarting. Drug-Induced Liver Injury [see Warnings and Precautions ( 5.10 )] Grade 3 ALT or higher in the absence of another cause 4 OR AST/ALT greater than 3 × upper limit normal (ULN) with bilirubin greater than 2 × ULN in the absence of another cause 4 Permanently discontinue. Non-hematological toxicities [see Adverse Reactions ( 6.1 )] Grade 2 Continue treatment at reduced dose by 1 dose level. Grade 3 1st occurrence Interrupt until Grade 2 or less, then resume at reduced dose by 1 dose level. No improvement within 7 days OR 2 nd or 3 rd occurrence Interrupt until Grade 2 or less, then resume at reduced dose by 2 dose levels. 4 th occurrence Interrupt until Grade 2 or less, then resume at reduced dose by 2 dose levels for HCC and RCC or 3 dose levels for DTC. Grade 4 Permanently discontinue. 1 Adverse reactions graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0). 2 If no recovery after 30 day interruption, discontinue treatment unless the patient is deriving clinical benefit. 3 If more than 2 dose reductions are required, permanently discontinue treatment. 4 In addition, any grade increased alkaline phosphatase in the absence of known bone pathology and Grade 2 or worse increased bilirubin; any 1 of the following: INR of 1.5 or greater, ascites and/or encephalopathy in the absence of underlying cirrhosis or other organ failure considered to be due to drug-induced liver injury. Table 3: Recommended Dosage Modifications for Dermatologic Toxicities Dermatologic Toxicity Grade Occurrence Sorafenib Tablets Dosage Modification Hepatocellular and Renal Cell Carcinoma Differentiated Thyroid Carcinoma Grade 2: Painful erythema and swelling of the hands or feet and/or discomfort affecting the patient’s normal activities 1 st occurrence Continue sorafenib tablets and consider topical therapy for symptomatic relief. If no improvement within 7 days, see below. Decrease sorafenib tablets to 600 mg daily. If no improvement within 7 days, see below. No improvement within 7 days at reduced dose OR 2 nd and 3 rd occurrence Interrupt sorafenib tablets until resolved or improved to Grade 0 to 1. Interrupt sorafenib tablets until completely resolved or improved to Grade1. When resuming treatment, decrease dose by 1 dose level. When resuming treatment, decrease dose by 1 dose level for 2 nd occurrence and 2 doses levels for 3 rd occurrence. 4 th occurrence Discontinue sorafenib tablets treatment. Grade 3: Moist desquamation, ulceration, blistering, or severe pain of the hands or feet, resulting in inability to work or perform activities of daily living 1 st occurrence Interrupt sorafenib tablets until resolved or improved to Grade 0 to 1 Interrupt sorafenib tablets until completely resolved or improved to Grade 1. When resuming treatment, decrease dose by 1 dose level. When resuming treatment, decrease dose by 1 dose level. 2 nd occurrence Interrupt sorafenib tablets until resolved or improved to Grade 0 to 1 Interrupt sorafenib tablets until completely resolved or improved to Grade 1. When resuming treatment, decrease dose by 1 dose level. When resuming treatment, decrease dose by 2 dose levels. 3 rd occurrence Discontinue sorafenib tablets treatment Following improvement of Grade 2 or 3 dermatologic toxicity to Grade 0 or 1 for at least 28 days on a reduced dose of sorafenib tablets, the dose of sorafenib tablets may be increased 1 dose level from the reduced dose. Approximately 50% of patients requiring a dose reduction for dermatologic toxicity are expected to meet these criteria for resumption of the higher dose and roughly 50% of patients resuming the previous dose are expected to tolerate the higher dose (that is, maintain the higher dose level without recurrent Grade 2 or higher dermatologic toxicity).
Warnings
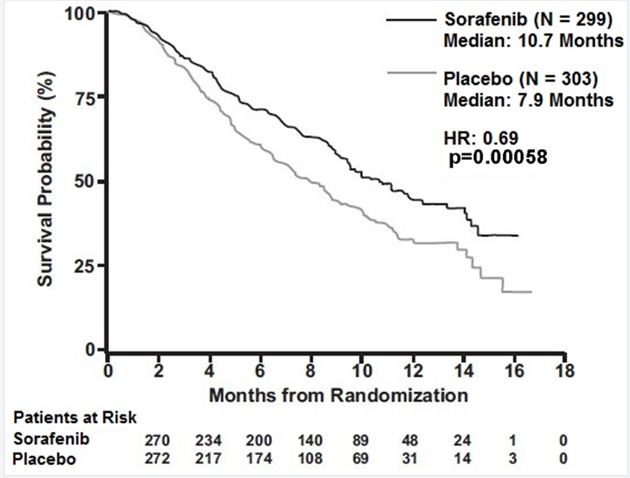
Cardiovascular Events: Consider temporary or permanent discontinuation of sorafenib tablets.(2.2, 5.1) Hemorrhage: Discontinue sorafenib tablets if needed. (5.2) Hypertension: Monitor blood pressure weekly during the first 6 weeks and periodically thereafter. Consider temporary or permanent discontinuation for severe or persistent hypertension despite antihypertensive therapy.(5.3) Dermatologic Toxicities: Interrupt and/or decrease dose. Discontinue for severe or persistent reactions, or if Stevens-Johnson syndrome and toxic epidermal necrolysis is suspected.(5.4) Gastrointestinal Perforation: Discontinue sorafenib tablets. (5.5) Risk of Impaired Wound Healing: Withhold sorafenib tablets for at least 10 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of sorafenib tablets after resolution of wound healing complications has not been established. (5.7) QT Prolongation: Monitor electrocardiograms and electrolytes in patients at increased risk for ventricular arrhythmias. Correct electrolytes. Interrupt if QTc greater than 500 msec or increases greater than 60 msec from baseline.( 2.2, 5.9, 12.2) Drug-Induced Liver Injury: Monitor liver function tests regularly; discontinue for unexplained transaminase elevations. (5.10) Embryo-Fetal Toxicity: Sorafenib tablets may cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.11, 8.1, 8.3) Impairment of Thyroid Stimulating Hormone Suppression (TSH) in DTC: Monitor TSH monthly and adjust thyroid replacement therapy in patients with thyroid cancer. (5.12) 5.1 Cardiovascular Events In the SHARP (HCC) study, the incidence of cardiac ischemia/infarction was 2.7% in sorafenib tablets-treated patients compared with 1.3% in those receiving placebo, and in the DECISION (DTC) study, the incidence of cardiac ischemia/infarction was 1.9% in the sorafenib tablets -treated group compared with 0% in patients receiving placebo. Patients with unstable coronary artery disease or recent myocardial infarction were excluded from this study. In multiple clinical trials, congestive heart failure has been reported in 1.9% of sorafenib tablets - treated patients (N=2276) [see Adverse Reactions (6.2)]. Consider temporary or permanent discontinuation of sorafenib tablets in patients who develop cardiovascular events [see Dosage and Administration (2.2)]. 5.2 Hemorrhage An increased risk of bleeding may occur following sorafenib tablets administration. In the SHARP (HCC) study, the rates of bleeding from esophageal varices (2.4% and 4%) and of bleeding with a fatal outcome from any site (2.4% and 4%) were similar in sorafenib tablets -treated patients and those receiving placebo, respectively. In the DECISION (DTC) study, bleeding was reported in 17.4% of sorafenib tablets -treated patients and 9.6% of those receiving placebo; however, the incidence of Grade 3 bleeding was similar (1% and 1.4%) in sorafenib tablets -treated patients and in those receiving placebo. If any bleeding necessitates medical intervention, consider permanent discontinuation of sorafenib tablets [see Dosage and Administration (2.2)]. Due to the potential risk of bleeding, treat tracheal, bronchial, and esophageal infiltration with local therapy prior to administering sorafenib tablets in patients with DTC. 5.3 Hypertension In the SHARP (HCC) study, hypertension was reported in 9.4% of sorafenib tablets -treated patients and 4.3% of patients receiving placebo. In the DECISION (DTC) study, hypertension was reported in 40.6% of sorafenib tablets -treated patients and 12.4% of patients receiving placebo. Hypertension was usually mild to moderate, occurred early in the course of treatment, and was managed with standard antihypertensive therapy. Permanent discontinuation due to hypertension occurred in 1 of 297 sorafenib tablets -treated patients in the SHARP (HCC) study, and 1 of 207 sorafenib tablets -treated patients in the DECISION (DTC) study. Monitor blood pressure weekly during the first 6 weeks of sorafenib tablets. Thereafter, monitor blood pressure and treat hypertension, if required, in accordance with standard medical practice. In cases of severe or persistent hypertension despite institution of antihypertensive therapy, consider temporary or permanent discontinuation of sorafenib tablets [see Dosage and Administration (2.2)]. 5.4 Dermatologic Toxicities Hand-foot skin reaction and rash represent the most common adverse reactions attributed to sorafenib tablets. Rash and hand-foot skin reaction are usually Grade 1 and 2 and generally appear during the first six weeks of treatment with sorafenib tablets. Permanent discontinuation of therapy due to hand-foot skin reaction occurred in 4 (1.3%) of 297 sorafenib tablets -treated patients with HCC, and 11 (5.3%) of 207 sorafenib tablets -treated patients with DTC. Management of dermatologic toxicities may include topical therapies for symptomatic relief, temporary treatment interruption and/or dose reduction of sorafenib tablets, or in severe or persistent cases, permanent discontinuation of sorafenib tablets [see Dosage and Administration (2.2)]. There have been reports of severe dermatologic toxicities, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). These cases may be life-threatening. Discontinue sorafenib tablets if SJS or TEN are suspected. 5.5 Gastrointestinal Perforation Gastrointestinal perforation has been reported in less than 1% of patients taking sorafenib tablets. In some cases this was not associated with apparent intra-abdominal tumor. In the event of a gastrointestinal perforation, permanently discontinue sorafenib tablets. 5.6 Increased Risk of Bleeding with Concomitant Us e of Warfarin Infrequent bleeding or elevations in the International Normalized Ratio (INR) have been reported in some patients taking warfarin while on sorafenib tablets. Monitor patients taking concomitant warfarin regularly for changes in prothrombin time (PT), INR or clinical bleeding episodes. 5.7 Risk of Impaired Wound Healing Impaired wound healing can occur in patients who receive drugs that inhibit the VEGF signaling pathway. Therefore, sorafenib tablets has the potential to adversely affect wound healing. Withhold sorafenib tablets for at least 10 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of sorafenib tablets after resolution of wound healing complications has not been established. 5.8 Increased Mortality Observed with Sorafenib Tablets Administered in Combination with Carboplatin/Paclitaxel and Gemcitabine/Cisplatin in Squamous Cell Lung Cancer In a subset analysis of two randomized controlled trials in chemo-naive patients with Stage IIIB-IV non-small cell lung cancer, patients with squamous cell carcinoma experienced higher mortality with the addition of sorafenib tablets compared to those treated with carboplatin/paclitaxel alone (HR 1.81; 95% CI 1. 19, 2.74) and gemcitabine/cisplatin alone (HR 1.22; 95% CI 0.82, 1.80). The use of sorafenib tablets in combination with carboplatin/paclitaxel is contraindicated in patients with squamous cell lung cancer. Sorafenib tablets in combination with gemcitabine/cisplatin are not recommended in patients with squamous cell lung cancer. The safety and effectiveness of sorafenib tablets has not been established in patients with non-small cell lung cancer. 5.9 QT Interval Prolongation Sorafenib tablets can prolong the QT/QTc interval. QT/QTc interval prolongation increases the risk for ventricular arrhythmias. Avoid sorafenib tablets in patients with congenital long QT syndrome. Monitor electrolytes and electrocardiograms in patients with congestive heart failure, bradyarrhythmias, drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics. Correct electrolyte
Contraindications
CONTRAINDICATIONS • Sorafenib tablets are contraindicated in patients with known severe hypersensitivity to sorafenib or any other component of sorafenib tablets. • Sorafenib tablets in combination with carboplatin and paclitaxel is contraindicated in patients with squamous cell lung cancer [see Warnings and Precautions ( 5.8 )]. • Sorafenib tablets are contraindicated in patients with known severe hypersensitivity to sorafenib or any other component of sorafenib tablets. ( 4 ) • Sorafenib in combination with carboplatin and paclitaxel is contraindicated in patients with squamous cell lung cancer. ( 4 )
Drug interactions
Strong CYP3A Inducers: Avoid strong CYP3A4 inducers.(7.1) 7.1 Effect of Other Drugs on Sorafenib Tablets Strong CYP3A4 Inducers The concomitant use of sorafenib tablets with rifampin, a strong CYP3A4 inducer decreased the mean AUC of sorafenib, which may decrease the antitumor activity [see Clinical Pharmacology (12.3)]. Avoid concomitant use of sorafenib tablets with strong CYP3A4 inducers, when possible, because these drugs can decrease the systemic exposure to sorafenib. Neomycin The concomitant use of sorafenib tablets with neomycin decreased the mean AUC of sorafenib, which may decrease the antitumor activity. Avoid concomitant use of sorafenib tablets with neomycin. The effects of other antibiotics on the pharmacokinetics of sorafenib have not been studied [see Clinical Pharmacology (12.3)]. 7.2 Concomitant Use of Warfarin The concomitant use of sorafenib tablets and warfarin may increase the risk of bleeding or increased the INR. Monitor INR and for clinical bleeding episodes in patients taking warfarin while receiving sorafenib tablets [see Warnings and Precautions (5.6)]. 7.3 Drugs That Prolong the QT Interval Sorafenib tablets are associated with QTc interval prolongation. Avoid coadministration of sorafenib tablets with medicinal products with a known potential to prolong QT/QTc interval [see Warnings and Precautions (5.9), Clinical Pharmacology (12.2)]. 8.7 Hepatic Impairment No dose adjustment is necessary for patients with mild or moderate hepatic impairment. The pharmacokinetics of sorafenib have not been studied in patients with severe (Child-Pugh C) hepatic impairment [see Clinical Pharmacology (12.3)].
Adverse reactions
The most common adverse reactions (≥20%) are diarrhea, fatigue, infection, alopecia, hand-foot skin reaction, rash, weight loss, decreased appetite, nausea, gastrointestinal and abdominal pains, hypertension, and hemorrhage. (6) To report SUSPECTED ADVERSE REACTIONS, contact Yabao Pharmaceutical Co., Ltd. Beijing at 914-656-3049 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch 6 ADVERSE REACTIONS The following clinically significant adverse reactions are discussed elsewhere in the labeling: Cardiovascular events [see Warnings and Precautions ( 5.1 )] Hemorrhage [see Warnings and Precautions ( 5.2 )] Hypertension [see Warnings and Precautions ( 5.3 )] Dermatologic toxicities [see Warnings and Precautions ( 5.4 )] Gastrointestinal perforation [see Warnings and Precautions ( 5.5 )] QT interval prolongation [see Warnings and Precautions ( 5.9 ) and Clinical Pharmacology( 12.2 )] Drug-induced liver injury [see Warnings and Precautions ( 5.10 )] Impairment of TSH suppression in DTC [see Warnings and Precautions ( 5.12 )] 6.1 Clinical Trials Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The data described reflect exposure to sorafenib tablets in 504 patients who participated in placebo-controlled studies in hepatocellular carcinoma N=297), or differentiated thyroid carcinoma (N = 207). The most common adverse reactions (≥20%), which were considered to be related to sorafenib tablets, in patients with HCC or DTC are diarrhea, fatigue, infection, alopecia, hand-foot skin reaction, rash, weight loss, decreased appetite, nausea, gastrointestinal and abdominal pains, hypertension, and hemorrhage. Hepatocellular Carcinoma Table 4 shows the percentage of patients in the SHARP (HCC) study experiencing adverse reactions that were reported in at least 10% of patients and at a higher rate in the Sorafenib Tablets -treated group than in those eceiving placebo. Table 4: Adverse Reactions Reported in at Least 10% of Patients and at a Higher Rate in Sorafenib Tablets Arm than the Placebo Arm–SHARP (HCC) Adverse Reaction 1 Sorafenib Tablets N = 297 Placebo N = 302 All Grades% Grade 3% Grade 4% All Grades % Grade 3% Grade 4% Any Adverse Reaction 98 39 6 96 24 8 Gastrointestinal Diarrhea 55 10 < 1 25 2 0 Anorexia 29 3 0 18 3 < 1 Nausea 24 1 0 20 3 0 Vomiting 15 2 0 11 2 0 Constipation 14 0 0 10 0 0 Constitutional symptoms Fatigue 46 9 1 45 12 2 Weight loss 30 2 0 10 1 0 Pain Pain, abdomen 31 9 0 26 5 1 Dermatology/skin Hand-foot skin reaction 21 8 0 3 < 1 0 Rash/desquamation 19 1 0 14 0 0 Alopecia 14 0 0 2 0 0 Pruritus 14 < 1 0 11 < 1 0 Dry skin 10 0 0 6 0 0 Hepatobiliary/pancreas Liver dysfunction 11 2 1 8 2 1 1Adverse reactions graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0). Hypertension was reported in 9% of patients treated with sorafenib tablets and 4% of those receiving placebo. Grade 3 hypertension was reported in 4% of sorafenib tablets -treated patients and 1% of those receiving placebo. Hemorrhage/bleeding was reported in 18% of those receiving sorafenib tablets and 20% of patients receiving placebo. The rates of Grade 3 and 4 bleeding were also higher in patients receiving placebo (Grade 3 – 3% sorafenib tablets and 5% placebo and Grade 4 – 2% sorafenib tablets and 4% placebo). Bleeding from esophageal varices was reported in 2.4% in sorafenib tablets -treated patients and 4% of patients receiving placebo. Renal failure was reported in <1% of patients treated with sorafenib tablets and 3% of patients receiving placebo. Clinical pancreatitis was reported in 1 of 297 sorafenib tablets -treated patients (Grade 2). The rate of adverse reactions (including those associated with progressive disease) resulting in permanent discontinuation was similar in both the sorafenib tablets -treated patients and those receiving placebo (32% of sorafenib tablets -treated patients and 35% of patients receiving placebo). Laboratory test abnormalities reported in SHARP are presented in Table 5. Table 5: Laboratory Test Abnormalities Reported in SHARP (HCC) Laboratory Parameter 1 Sorafenib Tablets N=297 Placebo N=302 All Grades(%) Grade 3 or 4(%) All Grades(%) Grade 3 or 4(%) Hypoalbuminemia 59 0 47 0 Elevated Lipase 40 9 37 9 Lymphopenia 47 NR 42 NR Thrombocytopenia 46 4 41 < 1 Elevated INR 42 4 34 2 Hypophosphatemia 35 11 11 2 Elevated Amylase 34 2 29 2 Hypocalcemia 27 2.4 15 1 Hypokalemia 10 < 1 6 < 1 1Laboratory parameters graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0). NR = not reported Differentiated Thyroid Carcinoma The safety of sorafenib tablets was evaluated in DECISION in 416 patients with locally recurrent or metastatic, progressive differentiated thyroid carcinoma (DTC) refractory to radioactive iodine (RAI) treatment randomized to receive 400 mg twice daily sorafenib tablets (n=207) or matching placebo (n=209) until disease progression or intolerable toxicity in a double-blind trial [see Clinical Studies (14.3)]. The data described below reflect a median exposure to sorafenib tablets for 46 weeks (range 0.3 to 135). The population exposed to sorafenib tablets was 50% male, and had a median age of 63 years. Dose interruptions for adverse reactions were required in 66% of patients receiving sorafenib tablets and dose reductions were required in 64% of patients. Adverse reactions that resulted in treatment discontinuation were reported in 14% of sorafenib tablets -treated patients compared to 1.4% of patients receiving placebo. Table 8 shows the percentage of DTC patients experiencing adverse reactions at a higher rate in sorafenib tablets -treated patients than in patients receiving placebo in the double-blind phase of the DECISION study. Grade 3 adverse reactions occurred in 53% of sorafenib tablets -treated patients compared to 23% of patients receiving placebo. Grade 4 adverse reactions occurred in 12% of sorafenib tablets-treated patients compared to 7% of patients receiving placebo. Table 8: Selected Adverse Reactions Occurring at a Higher Incidence in Sorafenib Tablets -Treated Patients [Between Arm Difference of ≥ 5% (All Grades)1 or ≥ 2% (Grades 3 and 4)] Adverse Reaction Sorafenib Tablets N = 207 Placebo N = 209 All Grades (%) Grades 3 and 4 (%) All Grades (%) Grades 3 and 4 (%) Skin and subcutaneous tissue disorders PPES 5 69 19 8 0 Alopecia 67 0 8 0 Rash 35 5 7 0 Pruritus 20 0.5 11 0 Dry skin 13 0.5 5 0 Erythema 10 0 0.5 0 Hyperkeratosis 7 0 0 0 Gastrointestinal disorders Diarrhea 68 6 15 1 Stomatitis 3 24 2 3 0 Nausea 21 0 12 0 Abdominal pain 2 20 1 7 1 Constipation 16 0 8 0.5 Oral pain 4 14 0.5 6 0 Vomiting 11 0 3 0 Investigations Weight loss 49 6 14 1 General disorders and administration site conditions Fatigue 41 5 20 1 Asthenia 12 0 7 0 Pyrexia 11 1 5 0 Vascular disorders Hypertension 6 41 10 12 2 Metabolism and nutrition disorders Decreased appetite 30 2 5 0 Nervous system disorders Headache 17 0 6 0 Dysgeusia 6 0 0 0 Musculoskeletal and connective tissue disorders Pain in extremity 15 1 7 0 Muscle spasms 10 0 3 0 Respiratory, thoracic and mediastinal disorders Dysphonia 13 0.5 3 0 Epistaxis 7 0 1 0 Neoplasms benign, malignant and unspecified Squamous cell carcinoma of skin 3 3 0 0 1 National Cancer Institute Common Terminology Criteria for Adverse Events Version 3.0 2 Includes the following terms: abdominal pain, abdominal discomfort, hepatic pain, esophageal pain, esophageal discomfort, abdominal pain lower, abdominal pain upper, abdominal tenderness, abdominal rigidity 3 Includes the following terms: stomatitis, aphthous stomatitis, mouth ulceration, mucosal inflammation 4 Includes the following terms: oral pain, oropharyngeal discomfort, glossitis, burning mouth syndrome, glossod
Mechanism of action
Mechanism of Action Sorafenib is a kinase inhibitor that decreases tumor cell proliferation in vitro. Sorafenib was shown to inhibit multiple intracellular (c-CRAF, BRAF and mutant BRAF) and cell surface kinases (KIT, FLT- 3, RET, RET/PTC, VEGFR-1, VEGFR- 2, VEGFR- 3, and PDGFR-ß). Several of these kinases are thought to be involved in tumor cell signaling, angiogenesis and apoptosis. Sorafenib inhibited tumor growth of HCC and DTC human tumor xenografts in immunocompromised mice. Reductions in tumor angiogenesis were seen in models of HCC upon sorafenib treatment, and increases in tumor apoptosis were observed in models of HCC and DTC. 12.2 Pharmacodynamics Cardiac Electrophysiology The effect of sorafenib tablets 400 mg twice daily on the QTc interval was evaluated in a multi-center, open- label, non-randomized trial in 53 patients with advanced cancer. No large changes in the mean QTc intervals (that is, >20 ms) from baseline were detected in the trial. After one 28-day treatment cycle, the largest mean QTc interval change of 8.5 ms (upper bound of two-sided 90% confidence interval, 13.3 ms) was observed at 6 hours post-dose on day 1 of cycle 2 [see Warnings and Precautions (5.9), Drug Interactions (7.3)]. 12.3 Pharmacokinetics Multiple doses of sorafenib tablets for 7 days resulted in a 2.5- to 7-fold accumulation compared to a single dose. Steady-state plasma sorafenib concentrations were achieved within 7 days, with a peak-to-trough ratio of mean concentrations of less than 2. The steady-state concentrations of sorafenib following administration of sorafenib tablets 400 mg twice daily were evaluated in DTC and HCC patients. Patients with DTC have mean steady-state concentrations that are 1.8-fold higher than patients with HCC. The reason for increased sorafenib concentrations in DTC patients is unknown. Mean C max and AUC increased less than proportionally beyond oral doses of 400 mg administered twice daily. Absorption After administration of sorafenib tablets, the mean relative bioavailability was 38–49% when compared to an oral solution. Following oral administration, sorafenib reached peak plasma levels in approximately 3 hours. Effects of Food With a moderate-fat meal (30% fat; 700 calories), bioavailability was similar to that in the fasted state. With a high-fat meal (50% fat; 900 calories), bioavailability was reduced by 29% compared to that in the fasted state. Distribution In vitro binding of sorafenib to human plasma proteins was 99.5%. Elimination The mean elimination half-life of sorafenib was approximately 25 to 48 hours. Me tabolism Sorafenib undergoes oxidative metabolism by hepatic CYP3A4, as well as glucuronidation by UGT1A9. Excretion Sorafenib accounted for approximately 70–85% of the circulating analytes in plasma at steady-state. Eight metabolites of sorafenib have been identified, of which 5 have been detected in plasma. The main circulating metabolite of sorafenib, the pyridine N-oxide that comprises approximately 9–16% of circulating analytes at steady-state, showed in vitro potency similar to that of sorafenib. Following oral administration of a 100 mg dose of a solution formulation of sorafenib, 96% of the dose was recovered within 14 days, with 77% of the dose excreted in feces and 19% of the dose excreted in urine as glucuronidated metabolites. Unchanged sorafenib, accounting for 51% of the dose, was found in feces but not in urine. Specific Populations A study of the pharmacokinetics of sorafenib indicated that the mean AUC of sorafenib in Asians (N=78) was 30% lower than in Whites (N=40). Sex and age do not have a clinically meaningful effect on the pharmacokinetics of sorafenib. Patients with Renal Impairment Mild (CLcr 50-80 mL/min), moderate (CLcr 30 - <50 mL/min), and severe (CLcr <30 mL/min) renal impairment do not affect the pharmacokinetics of sorafenib [see Use in Specific Populations (8.6)] . Patients with Hepatic Impairment Mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment do not affect the pharmacokinetics of sorafenib [see Use in Specific Populations (8.7)] . Drug Interactions Studies Effect of Strong CYP3A4 Inhibitors on Sorafenib : Ketoconazole, a strong inhibitor of CYP3A4 and P- glycoprotein, administered at a dose of 400 mg once daily for 7 days did not alter the mean AUC of a single oral dose of sorafenib tablets 50 mg in healthy subjects. Effect of Strong CYP3A4 Inducers on Sorafenib : Concomitant use of sorafenib tablets with rifampin administered at a dose of 600 mg once daily for 5 days with a single oral dose of sorafenib tablets 400 mg in healthy volunteers resulted in a 37% decrease in the mean AUC of sorafenib. Effect of Neomycin on Sorafenib: Neomycin administered as an oral dose of 1 g three times daily for 5 days decreased the mean AUC of sorafenib by 54% in healthy subjects administered a single oral dose of sorafenib tablets 400 mg. Effect of Sorafenib on Other Drugs: Sorafenib tablets 400 mg twice daily for 28 days did not increase the systemic exposure of concomitantly administered midazolam (CYP3A4 substrate), dextromethorphan (CYP2D6 substrate), and omeprazole (CYP2C19 substrate) [see Clinical Pharmacology (12.3)] . Drugs that Increase Gastric pH: The aqueous solubility of sorafenib is pH dependent, with higher pH resulting in lower solubility. However, omeprazole, a proton pump inhibitor, administered at a dose of 40 mg once daily for 5 days, did not result in a clinically meaningful change in sorafenib single dose exposure. In Vitro Studies Sorafenib competitively inhibited CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 in vitro. However, sorafenib tablets 400 mg twice daily for 28 days with substrates of CYP3A4, CYP2D6 and CYP2C19 did not increase the systemic exposure of these substrates [see Drug Interactions (7.3)] . Sorafenib did not increase CYP1A2 and CYP3A4 activities, suggesting that sorafenib is unlikely to induce CYP1A2 or CYP3A4 in humans. Sorafenib inhibits glucuronidation by UGT1A1 and UGT1A9 in vitro . Sorafenib tablets could increase the systemic exposure of concomitantly administered drugs that are UGT1A1 or UGT1A9 substrates. Sorafenib inhibited P-glycoprotein in vitro . Sorafenib tablets could increase the concentrations of concomitantly administered drugs that are P-glycoprotein substrates.
Available forms (2)
NDC examples
51990-20124979-71543598-458
Treats these conditions
Source: openFDA + RxNorm · 2026
Look up another medication